QuickParanoid - A Tool for Ortholog Clustering
-
by Taekyung Kim and Sungwoo Park
Introduction
QuickParanoid is a suite of programs for automatic ortholog clustering and analysis. It takes as input a collection of files produced by InParanoid and finds ortholog clusters among multiple species. For a given dataset, QuickParanoid first preprocesses each InParanoid output file and then computes ortholog clusters. It also provides a couple of programs qa1 and qa2 for analyzing the result of ortholog clustering.QuickParanoid is similar to MultiParanoid and OrthoMCL in functionality, but is much faster. For example, it takes only 199.56 seconds on an Intel 2.4Ghz machine with 1 gigabyte memory to process a dataset of 120 species (which contains 14403218 entries in 120 * 119 / 2 = 7140 InParanoid output files of a total size of 365.38 megabytes). In comparison, MultiParanoid on the same machine fails to process a dataset of 20 species (which contains 319368 entries), and OrthoMCL fails to process a dataset of 60 species (which contains 3245394 entries). The accuracy is also comparable with MultiParanoid and OrthoMCL. For example, for a dataset of 3 species, QuickParanoid finds 135 clusters in the manually curated data consisting of 221 clusters while MultiParanoid and OrthoMCL find 137 clusters and 98 clusters, respectively.
Here is the result of testing the speed and memory usage of the three programs using eight different datasets. All experiments were performed on an Intel 2.4Ghz machine running Debian Linux 2.6.21-6 with 1 gigabyte memory. Memory usage for MultiParanoid and OrthoMCL was measured using top. Note that in the experiment with 120 species, QuickParanoid finds a cluster consisting of sequences from all 120 species!
| Number of species | Dataset size | Number of entries in the dataset | QuickParanoid [summary]
(running time) (memory usage) (number of clusters found) |
MultiParanoid
(running time) (memory usage) (number of clusters found) |
OrthoMCL
(running time) (memory usage) (number of clusters found) |
Number of clusters found by QuickParanoid and MultiParanoid | Number of clusters found by QuickParanoid and OrthoMCL |
| 5 | 0.38 Mbytes | 14664 | 0.15 seconds 600 Kbytes 2208 clusters |
2.03 seconds 38 Mbytes 2293 clusters |
35 seconds 31.00 Mbytes 2787 clusters |
2091 clusters | 1372 clusters |
| 10 | 1.86 Mbytes | 71035 | 0.51 seconds 1584 Kbytes 3034 clusters |
48.78 seconds 140 Mbytes 3218 clusters |
175 seconds 61.75 Mbytes 4466 clusters |
2737 clusters | 1882 clusters |
| 15 | 4.24 Mbytes | 164173 | 1.09 seconds 3117 Kbytes 4242 clusters |
5107.86 seconds 314 Mbytes 4515 clusters |
600 seconds 112.45 Mbytes 6849 clusters |
3767 clusters | 2751 clusters |
| 20 | 8.19 Mbytes | 319368 | 2.75 seconds 4764 Kbytes 4934 clusters |
∞
- - |
1150 seconds 186.50 Mbytes 8477 clusters |
- | 3259 clusters |
| 40 | 35.14 Mbytes | 1407029 | 13.10 seconds 28655 Kbytes 9003 clusters |
∞
- - | 13513 seconds 722.75 Mbytes 18686 clusters |
- | 5539 clusters |
| 60 | 81.48 Mbytes | 3245394 | 40.32 seconds 94425 Kbytes 18830 clusters |
∞
- - |
- | - | - |
| 90 | 225.07 Mbytes | 8865949 | 84.12 seconds 245024 Kbytes 27199 clusters |
∞
- - |
- | - | - |
| 120 | 365.38 Mbytes | 14403218 | 199.56 seconds 335972 Kbytes 29379 clusters |
∞
- - |
- | - | - |
Here is the result of testing the accuracy of the three programs using a dataset of three species (human, fly, worm) for which manually curated data are available. Each entry in the table denotes the number of clusters.
| Manually curated data (A) | QuickParanoid (B) | MultiParanoid (C) | OrthoMCL (D) | A ∩ B | A ∩ C | A ∩ D | A ∩ B ∩ C | A ∩ B ∩ D |
| 221 | 5620 | 5722 | 5635 | 135 | 137 | 98 | 135 | 92 |
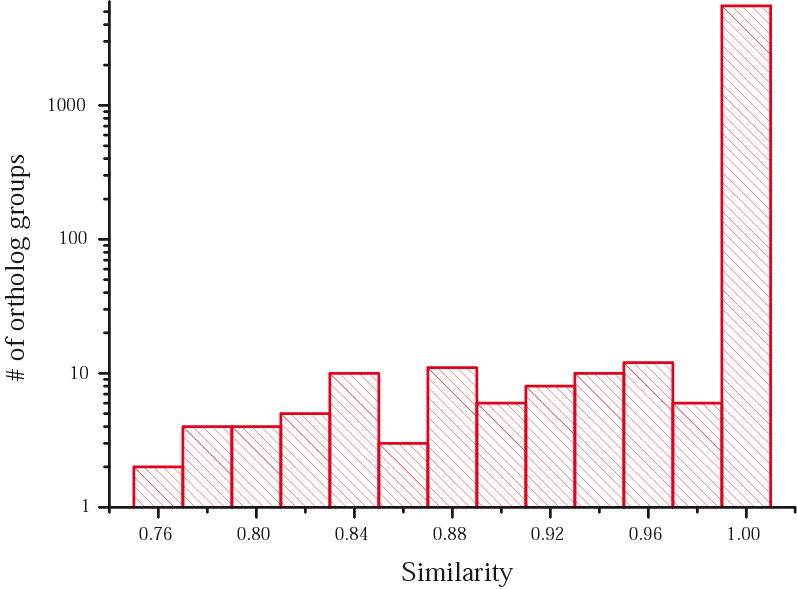
Among 5620 clusters found by QuickParanoid, 98.52% (5537 clusters) exactly match those found by MultiParanoid. The following graph shows the distribution of clusters found by QuickParanoid against their similarity with those found by MultiParanoid (in logarithmic scale). A similarity value p means that p of sequences in a cluster found by QuickParanoid are included in some similar cluster found by MultiParanoid. p = 1.0 means that exactly the same cluster is found both by QuickParanoind and by MultiParanoid.
Installation
- QuickParanoid requires GNU gcc and make in the Unix environment.
We have tested it only on Debian Linux 2.6.21-6 and Cygwin,
but any programming environment with GNU gcc and make should be fine.
- Download
QuickParanoid.tar.gz
or
QuickParanoid.zip, and uncompress it in your installation directory.
- Run make qa in the installation directory to see if GNU gcc and make are available.
[gla@plquad:40 ] make qa g++ -o qa1 qa1.cpp gcc -c hashtable.c -o hashtable.o g++ -o qa2 hashtable.o qa2.cpp
If successful, you are ready to use QuickParanoid. If not, make sure that both gcc and make are installed on your system, and edit Makefile appropriately (two variables CC and CPP in it). - If your compiler complains that strcpy() and strcmp() are not declared, import "string.h" in your source code.
Quick guide to QuickParanoid
- Follow the instruction to install QuickParanoid.
- Download a dataset of three species fly-human-worm.tar.gz or
fly-human-worm.zip
in the same directory that QuickParanoid is installed, and uncompress it.
-
Run qp and follow the instruction:
[gla@plquad:43 ] qp ===================================================== QuickParanoid ===================================================== Dataset directory [default = "." (current directory)]: fly-human-worm Data file prefix [default = "sqltable."]: Data file separator [default = "-"]: Configuration file [default = "fly-human-worm/config"]: Executable file prefix [default = "test"]: Generating a header file..... Updating Makefile..... Generating executable files...... g++ -o dump dump.cpp ./dump fly-human-worm/config Reading the config file Reading the data files ... g++ -o gen_header gen_header.cpp ./gen_header fly-human-worm/config __ortholog.h Reading the config file Reading the data files Opening fly-human-worm/sqltable.fly2k-human2k Opening fly-human-worm/sqltable.fly2k-worm2k Opening fly-human-worm/sqltable.human2k-worm2k Generating structure definitions Generating functions Generating species Generating sequences gcc -c ortholog.c -o ortholog.o gcc -c hashtable_itr.c -o hashtable_itr.o gcc -o test hashtable.o hashtable_itr.o ortholog.o qp.c -lm gcc -o tests qps.c Done. Run test to perform ortholog clustering. Run tests to see the dataset size and the number of entries. - Run the generated programs, redirecting their output to a text file if necessary:
[gla@plquad:44 ] test > result.txt [gla@plquad:45 ] tests Dataset size in bytes: 1404502 Number of entries in the dataset: 44752
- In the above case, result.txt contains the result of ortholog clustering on the three species. It uses the same output format that MultiParanoid uses. [result.txt]
Usage
-
QuickParanoid builds an executable program specialized for each experiment of ortholog clustering.
First you need a dataset produced by InParanoid which may be stored in any working directory.
A dataset should have pairwise InParanoid output files between all species to be analyzed.
For analyzing N species, you need a dataset of N (N-1)/2 InParanoid output files.
(The rule for naming these files is explained later.)
Each line in the standard InParanoid output format consists of (1) cluster ID number; (2) InParanoid score; (3) species name; (4) seed score; (5) sequence name, as in the following example:
3 4425 fly2k 1.000 gi7303993
Optionally each line may contain a bootstrap value in an extra column after the sequence name, as in the following example:11 3607 ensMONDO.fa 1.000 ENSMODP00000019772 100%
Bootstrap values are not used by QuickParanoid and are ignored. -
Create a configuration file containing the names of species in the dataset.
List the name of each species in a separate line in the configuration file.
For example, an analysis of the three species fly2k, human2k, and worm2k
would use three lines as shown below:
[gla@plquad:49 ] cat fly-human-worm/config fly2k human2k worm2k
You may list the names of species in any order as long as you provide a dataset of N (N-1)/2 files for N species. The order of species does not affect the result of ortholog clustering. The only exception is that it may change the type of tree conflict (different-by-numbers or different-by-names) for those clusters with both types of tree conflicts, since QuickParanoid reports whichever type of tree conflict is detected first. -
Run qp (QuickParanoid) and provide all requested parameters.
For each parameter, a default value is displayed which is taken if the user presses just the enter key.
QuickParanoid assumes that all data files reside in the dataset directory and that every data file in it has a name concatenating the data file prefix, a species name, the data file separator, and another species name in that order. It also assumes that a configuration file can be located. All executable files begin with the executable file prefix. For example,
Dataset directory [default = "." (current directory)]: fly-human-worm Data file prefix [default = "sqltable."]: Data file separator [default = "-"]: Configuration file [default = "fly-human-worm/config"]: Executable file prefix [default = "test"]:
the data files reside in directory fly-human-worm, and sqltable.fly2k-human2k is the data file corresponding to species fly2k and human2k; a configuration file fly-human-worm/config is used, and two programs test and tests are generated. For the sake of parsing file names correctly, no species name should contain the data file separator.For each pair of species, the dataset must have at least one InParanoid output file. For example, if data file prefix is sqltable. and data file separator is -, two species A and B must have at least one of InParanoid output files sqltable.A-B and sqltable.B-A.
-
qp first preprocesses the entire dataset.
An executable program dump is generated, and
for each data file File in the dataset, a new file File_c
is created in the working directory.
For example,
a data file sqltable.fly2k-human2k generates a new file sqltable.fly2k-human2k_c.
(Two intermediate files __ortholog.h and gen_header are created in the installation directory.)
- Run the generated programs to perform ortholog clustering on those species specified in config,
or to see dataset size and the number of entries.
The result of ortholog clustering is displayed in the MultiParanoid output format.
Typically you would redirect the output to a text file as shown below:
[gla@plquad:44 ] test > result.txt [gla@plquad:45 ] tests Dataset size in bytes: 1404502 Number of entries in the dataset: 44752
- qa1 and qa2 are two helper programs.
qa1 analyzes the result of ortholog clustering, and
qa2 compares two different results of ortholog clustering from the same configuration file.
Run these programs with no argument to see their usage.
Examples of using these programs are:
[gla@plquad:50 ] qa1 Usage: qa1
The output of qa2 is self-explanatory. [analysis1.txt, analysis2.txt]
Datasets
- Dataset of three species fly2k, human2k, and worm2k:
fly-human-worm.tar.gz or
fly-human-worm.zip.
Uncompressing these files creates a subdirectory fly-human-worm. Manual.out is the manually curated data from the three species. It contains a total of 221 clusters. You may ignore the fourth column (is_seed_ortholog) to the last column (tree_conflict).
This dataset is provided by the authors of MultiParanoid (Andrey Alexeyenko, Ivica Tamas, Gang Liu, and Erik L.L. Sonnhammer (2006). Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics 22: e9-e15).
- Dataset of 120 species:
120species.tar.gz (179 megabytes in size; 755 megabytes when uncompressed).
Uncompressing the tar file creates a subdirectory 120species. It contains 8 configuration files config.5, config.10, config.15, config.20, config.30, config.60, config.90, and config.120. The suffix of each configuration file indicates the number of species in it.
This dataset was built from data available at NCBI GenBank (ftp://ftp.ncbi.nlm.nih.gov/genomes) using InParanoid.
Extending QuickParanoid
QuickParanoid preprocesses InParanoid output files to replace all string operations by much faster integer operations. If you wish to write your own program for analyzing InParanoid output files (e.g., another ortholog clustering program), you can use our code for preprocessing InParanoid output files so that you can concentrate on implementing your algorithm rather than handling input/output. This section explains how to use our code when writing such a program.If you follow the instruction in the Usage section to specify the dataset directory, the data file prefix, etc., a new header file __ortholog.h is created. Part of __ortholog.h that declares structures for reading InParanoid output files is as follows:
// sequence ---------------------------------------
typedef struct{
int species_id;
double seed;
int sequence_id;
} sequence;
// cluster ----------------------------------------
typedef struct{
int score;
int num_of_sequences;
sequence *sequences;
} cluster;
// dataFile ----------------------------------------
// invariant: species_id1 must be prior to species_id2 in the species table.
typedef struct{
int species_id1;
int species_id2;
int num_of_clusters;
cluster *clusters;
} dataFile;
//-------------------------------------------------------
// load_dataFile takes a pair of species ids and a dataFile
// and parses and loads the data file of two species.
// invariant: species_id1 < species_id2
void load_dataFile(int species_id1, int species_id2, dataFile* data);
// free_dataFile takes a pointer of a datafile and frees all memory.
void free_dataFile(dataFile* data);
- A dataFile structure is created as the result of reading an InParanoid output file. It stores an array of cluster structures, each of which describes a specific ortholog cluster.
- A cluster structure stores stores an array of sequence structures, each of which describes a specific sequence.
- A sequence structure records a sequence number (sequence_id), a species number (species_id), and its seed score.
-
To read an InParanoid output file,
you need two species numbers (of type int) and invoke load_dataFile.
The following code reads an InParanoid output file for species numbers s1 and s2
and stores the result in structure d:
d = (dataFile*)malloc(sizeof(dataFile)); load_dataFile(s1, s2, d);
// num of species
#define NUM_OF_SPECIES 3
// species id
// invariant: species id starts at 0.
#define _fly2k 0
#define _human2k 1
#define _worm2k 2
// species table
static const int species_table [] =
{_fly2k, _human2k, _worm2k};
// species names
// usage: species_names [species id]
static const char* species_names [] =
{"fly2k", "human2k", "worm2k"};
// num of sequence
#define NUM_OF_SEQUENCES 28494
// sequence names
// usage: sequence_names [sequence id]
char sequence_names[NUM_OF_SEQUENCES][15];
- NUM_OF_SPECIES is the total number of species in the entire dataset.
- species_names stores species names in such a way that species_names[i] returns the species name corresponding to species i.
- NUM_OF_SEQUENCES is the total number of sequences in the entire dataset.
- sequence_names stores sequence names in such a way that sequence_names[i] returns the sequence name corresponding to sequence i.
Your program should include __ortholog.h. An easy way to do this is to include the following two lines:
#include "qp.h" #include INTERMEDIATE_HEADER_FILE
You can compile your program in the same way that qp.c is compiled. If your program is foo.c, you can compile it as follows:
gcc hashtable.o hashtable_itr.o ortholog.o foo.c -lmThat's it!
Email us at
 .
.
Programming Language Laboratory Department of Computer Science and Engineering Pohang University of Science and Technology Republic of Korea